

Adaptation in Real Time:
Experimental Evolution with Yeast
Experimental evolution can help us to test hypotheses about evolution in controlled environments. Microbial systems can be especially powerful for these methods because their large population sizes and fast generation times allow us to develop quantitative insights into evolutionary processes. We can move from studying isolated examples and move towards generating probability distributions.

Lineage trajectories from our first evolution experiment (AKA Levy-Blundell experiment) with high-throughput barcoding. Lineages are colored according to the probability that they contain an established beneficial mutation. Adapted from Levy et al, 2015.
The Petrov lab (in collaboration with the Sherlock and Fisher labs) previously contributed to the development of high-throughput lineage tracking methods using barcodes that allow us to evolve and measure the fitness of hundreds of thousands of lineages in parallel. The group continues to use these, and similar methods, to empirically test fundamental questions from population genetics, systems biology, and, increasingly, eco-evolutionary theory.
Currently, we focus on a number of questions related to: i) the structure of the genotype-phenotype-fitness map; ii) how trade-offs constrain evolution; iii) the properties of evolution in fluctuating environments.
Genotype-Phenotype-Fitness Map
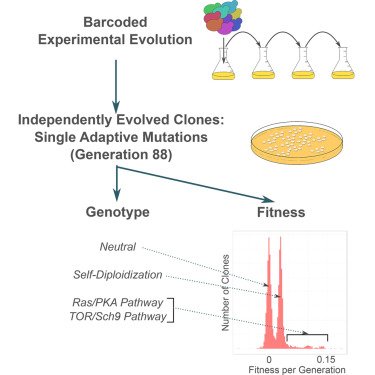
Isolation of adaptive lineages from the Levy-Blundell evolution experiment. Adapted from Venkataram et al, 2017.
Using barcode lineage tracing isolated hundreds of adaptive clones from an evolutionary experiment in the limited glucose environment. Each of these clones contained just a single adaptive mutation which we were able to identify by whole genome sequencing. We inferred that most were in two signaling pathways, RAS/PKA and Tor/Sch9. We were also able to precisely measure the fitness effects of individual clones using barcodes - the adaptive clones generated ~25% to 125% in fitness benefit per cycle indicating that adaptive mutations of very large effect are common. This paper defined possibly the first comprehensive genotype to fitness map of adaptation.
The ability to build the genotype-to-fitness map is an important first step. The next key step is to understand the full phenotypic effects of the adaptive mutations and what makes them adaptive. The problem here is both conceptual and technical. While we have means now to measure phenotype at high throughput, the number of phenotypes we could measure is virtually infinite. In addition, if a mutation leads to a change of expression of hundreds of genes, should we consider each change of expression of each gene as a separate phenotype? And how do we measure the effect of each phenotype on fitness?
Instead of measuring phenotype directly, we focused on the measurement of how fitness of individual mutants varies across environments. This genotype by environment variation in fitness must be related to phenotypic effects of mutations and gives us access to the space of fitness-related phenotypes. In the first such study we measured fitness of all adaptive mutations across environments that varied systematically in the time spent in fermentation, respiration, and stationary condition. We discovered that the strongest adaptive mutations in the RAS/PKA pathway obtained most of their benefit in respiration, which was surprising given that there is virtually no growth during respiration! We further determined that while the benefits do accrue during respiration they are only realized during the shortened lag phase of the next cycle. The ability of microbial populations to adapt using such “memory” is a novel finding and emphasizes that fitness should be measured per cycle across a number of cycles and not per generation as is commonly done.

The ‘fitness-relevant modularity’ model of adaptation. Adaptive mutations may collectively (and individually) affect many phenotypes, but only a small number of phenotypes may matter to fitness in the local environment (those indicated by black squares with thick arrows pointing to fitness), whereas other phenotypes may make very small contributions to fitness (those indicated by the gray squares and thin, dashed lines leading to fitness). The contribution of each phenotype to fitness changes depending on the environment. Adapted from Kinsler at al, 2020.
We have also pursed a more abstract approach in which we measure fitness across a very large number of random environments and infer the structure of the phenotypic space implicit in the structure of fitness values across all measurements. Using this approach we discovered that adaptive mutations change only a small number of fitness-relevant phenotypes in the evolving (glucose-limited) condition. Intriguingly, we also find that some of the phenotypic effects that are marginally important near the evolution condition become crucially important in distant environments. We argue that in this way directional selection can at times generate rather than destroy diversity. Overall, these studies demonstrate how we can use large numbers of individual adaptive mutations and precise measurements of fitness across environments to help understand the structure of phenotypic effects of adaptive mutations relevant to fitness.
We are continuing this work expanding to other environments and also using other computational approaches in an attempt to get to the underlying phenotypic structure that can be mapped to “real” measurable phenotypes and yield mechanistic understanding.
Tradeoffs and mapping of Pareto fronts
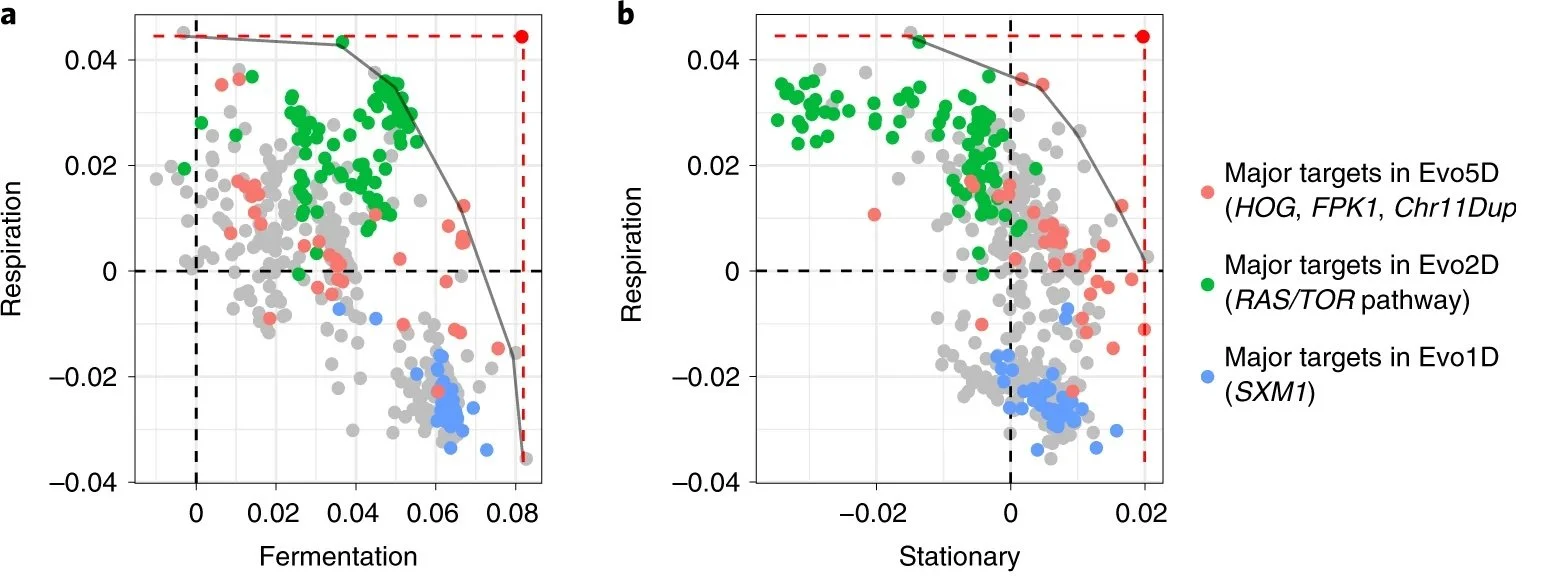
Pareto optimality fronts for respiration-fermentation (a) and respiration-stationary phase (b) discovered in the first-step evolution mutants. For each pair of performances, adaptive clones are plotted and colored according to their molecular basis. Each dot represents a clone. The large red dots represent the optimum phenotypes, achieving the upper limits (dashed lines) of each pair of performances. The grey curves, defined by the convex hull algorithm, represent putative Pareto optimality fronts. Adapted from Li et al, 2019.
Tradeoffs are commonly assumed in evolutionary theory but they are surprisingly hard to detect. In experimental evolution in particular, it is common to find adaptive mutations that improve all the measured traits seemingly implying lack of tradeoffs. We surmised that the reason for this pattern is that not all traits relevant to the organism are measured in general and certainly not in the laboratory setting. In this case one might be able to detect tradeoffs by looking at the largest effect mutations improving individual traits and detecting the absence of mutations that can improve multiple traits simultaneously to the same large effect. We used barcoding to carry out experimental evolution in environments that emphasized fermentation, respiration, and stationary stage fitness to different extents and used the large number of resulting first-step adaptive mutations to delineate tradeoffs in the form of the Pareto front even for the first step of evolution.
We are continuing this work by studying longer adaptive walks and attempting to understand the extent to which the observations of apparent tradeoffs and Pareto fronts are driven by the wiring of the cells and the consequent availability of specific adaptive mutations or by the “true” biophysical tradeoffs that arise given the way the organism is build in the physical sense.
Evolution in heterogeneous and fluctuating environments

An example of environmental memory common for adaptive mutants that evolved in a fluctuating environment. On the left: plotting fitness of the mutant pool in a fluctuating environment against the null hypothesis of the average of their fitness in the static environments reveals many cases of non-additivity, or vialotation from the 1:1 line. On the right: the frequency trajectory of an individual mutant (denoted by the magenta point on the left graph) shows an extreme example of non-additive fitness: for this mutant, fitness components of individual environments are opposite of those predicted by static environments. Adapted from Abreu et al, 2023.
One of the ways in which microbial experimental evolution studies fail to replicate natural evolution is the unnatural simplicity of environmental challenges in the experimental setting. We are investigating how the patterns of adaptation shift as the environmental challenges become progressively more complex. One of the key directions for us include the questions about
evolution dynamics of generalists and specialists in complex versus more simple environments,
whether the fitness in complex environments can be predicted easily from the fitness in the component simple environments,
whether evolution in complex and spatially distinct environments can drive coexistence of strains that specialize to the component environments.
Use of high-throughput genomic engineering: CRISPEY
While much of our work focuses on adaptive mutations that arise during experimental evolution, it is important to compare their properties with those of random mutations and polymorphisms segregating in natural populations. We are collaborating with the Fraser lab in implementing the high-throughput and efficient genome editing method CRISPEY in our lab. As the first step, we are measuring fitness and phenotypic effects of ~20,000 polymorphisms in the RAS/PKA pathway that we detected using the 1000 yeast genomes data and comparing their fitness and phenotypic properties with the adaptive mutations that arise in the RAS/PKA pathways in our experimental evolution in limited glucose.
Experimental evolution in the community context

During wine fermentation, the composition of microbial communities changes dramatically, yet the order of succession of yeast species is highly reproducible. This makes wine fermentation an excellent model system for studying evolution in a community context.
The vast majority of microbial evolution experiments have involved a single species adapting to a novel laboratory environment. In contrast, most microbes live in highly diverse ecosystems in which they have already undergone a long history of selection. Our lab is interested in whether the insights obtained from these single-species experiments extend to more natural community contexts or whether new phenomena emerge as one scales up the complexity. In order to bridge the gap between low- and high-diversity ecosystems, we have recently begun exploring communities of yeast associated with alcoholic fermentation, and, in particular, those involved in wine fermentation (with the help of Ignacio Belda at the Complutense University of Madrid). Using this new ‘in-vino’ system we are performing high-throughput evolution experiments with barcoded yeast adapting to communities of ‘intermediate’ complexity. We plan to use this system to quantify the impact of ecological complexity on the predictability of genomic evolution, on selection for intraspecific genetic variation and on the structure of fitness landscapes.